Clarifying the Molecular Basis of Cobalamin (Vitamin B12) Neurotrophism
G Scalabrino
DOI10.4172/2472-1913.100014
G Scalabrino*
Department of Biomedical Sciences, Laboratory of Neuropathology, University of Milan, 20133 Milan, Italy
- *Corresponding Author:
- G Scalabrino
M.D, Department of Biomedical Sciences
Laboratory of Neuropathology, University of Milan, 20133 Milan, Italy
Tel: +39 02 503111
E-mail: giuseppe.scalabrino@unimi.it
Received date: April 22, 2016; Accepted date: April 22, 2016; Published date: May 02, 2016
Citation: Scalabrino G. Clarifying the Molecular Basis of Cobalamin (Vitamin B12) Neurotrophism. J Headache Pain Manag. 2016, 1:2.
Copyright: © 2016 Scalabrino G. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Editorial
I have spent more than twenty-five years clarifying the molecular basis of the well-known neurotrophic effect of cobalamin (Cbl), more commonly known as vitamin B12. My studies have mainly concentrated on rat spinal cord (SC), the part of the central nervous system (CNS) that is most severely affected by Cbl deficiency both in humans and rats. The results obtained are described below in the order of the chronology and logical development of the research itself.
Identification of a new Cbl-deficient (Cbl-D) experimental model
I first identified a new experimental model reflecting all of the main neurological and haematological abnormalities characterising chronic Cbl deficiency in humans: the totally gastrectomised (TGX) rat. TGX rats become deficient in intrinsic factor immediately after total gastrectomy (TG) and Cbl-D over time, thus providing a surgical paradigm of pernicious anaemia [1, 2]. These rats develop the typical lesions of human subacute combined degeneration (i.e., Cbl-D neuropathy) in the CNS and peripheral nervous system (PNS): spongy vacuolation of SC white matter, CNS astroglial “activation”, and endoneural oedema, together with the “activation” of Schwann cells in the PNS. It was found that all of these pathological changes were reversed by the chronic post-TG administration of Cbl, thus suggesting that the damage is specifically related to the rats’ permanent Cbl-D status. No signs of demyelination or remyelination were found in either the CNS or PNS. As TG is a surgical procedure that leads to various severe consequences in rat, the morphological and biochemical findings were always compared with those observed in otherwise normal (ON) rats fed a Cbl-D diet, and there was a constant correspondence between the two experimental models [1, 2].
Identification of Cbl-regulated cytokines and growth factors
We next demonstrated that the severity of the neuropathological features of SC white matter in TGX rats did not correlate with serum or tissue accumulation of methylmalonic acid and homocysteine: i.e., there was no substantial increase in the severity of the SC white matter spongy vacuolation over time as the metabolites related to the failure of the Cbl-dependent enzymes (L-methylmalonyl-CoA mutase and homocysteine methyltransferase) accumulated in the SC, kidneys, liver and serum [2]. On the basis of this finding, we hypothesised that, rather than the simple withdrawal of the vitamin, deranged CNS production of some cytokines and/or neurotrophic factors might be a key element in the pathogenesis of Cbl-D central neuropathy in TGX rats. This hypothesis was supported by the fact that: a) different CNS cell types (mainly astrocytes) become “activated” after various CNS injuries and thus modify the synthesis and release of cytokines and/or growth factors; and b) optical and electron microscopy show that CNS glial cells (especially astrocytes) are severely affected by Cbl deficiency [1,2].
It was subsequently demonstrated that: a) Cbl down-regulates the level of tumour necrosis factor(TNF)-α in the SC of TGX rats; b) intracerebroventricular (i.c.v.) microinjections of agents antagonising the production and/or effects of TNF-, such as anti-TNF-α antibodies (Abs), interleukin(IL)-6, and transforming growth factor-β1, largely prevent spongy vacuolation in SC white matter of TGX rats; c) repeated i.c.v. microinjections of TNF-α cause spongy vacuolation in the SC white matter of ON rats that is similar to that observed in Cbl-D rats; d) Cbl down-regulates rat cerebrospinal fluid (CSF) but not serum levels of the soluble (s) CD40:sCD40 ligand (L) dyad, which belongs to the TNF-α:TNF- α-receptor superfamily; e) Cbl administration or inactivation by anti-CD40 Abs to TGX rats is accompanied by the normalisation of the myelin ultrastructure of SC white matter; f) Cbl up-regulates CSF levels of epidermal growth factor (EGF) and its synthesis in neurons and CNS glia; g) repeated i.c.v. microinjections of EGF into TGX rats are as effective as Cbl in significantly reducing the severity of spongy vacuolation in SC white matter even if their Cbl-D status remains unchanged; h) repeated i.c.v. microinjections of anti-EGF Abs cause spongy vacuolation in the SC white matter of ON rats that is similar to that observed in Cbl-D rats; i) Cbl up-regulates rat CSF levels of IL-6 (but not vasoactive intestinal peptide, somatostatin, or leptin); j) Cbl down-regulates the levels of nerve growth factor (NGF) by decreasing its synthesis in rat SC and its levels in rat CSF; k) although the Cbl deficiencyinduced peak in SC NGF synthesis occurs after the appearance of neuropathological lesions in the SC white matter of TGX rats, repeated i.c.v. microinjections of anti-NGF Abs shortly after TG substantially prevents the onset of such lesions; and l) Cbl downregulates transcobalamin receptor levels in different rat organs (including SC) [2-4].
We then hypothesised that the serum and CSF of adult patients with clinically confirmed severe Cbl deficiency might show a similar imbalance in TNF-α and EGF levels to that observed in the CNS and/or CSF of Cbl-D rats. It was demonstrated that there is such an imbalance in the serum of patients with pernicious anemia (PA), but not in those with severe iron-deficient anaemia, and that this imbalance can be rectified by Cbl-replacement therapy. This imbalance is also present in the CSF of patients with subacute combined degeneration (SCD), whose TNF-α levels are abnormally high and EGF levels abnormally low [3, 4].
All of the above indicated that the severe CNS damage caused by chronic Cbl deficiency in rat is due to a shift in the physiological equilibrium of CNS cytokines and growth factors in favour of myelinotoxic TNF-α and NGF, and against myelinotrophic EGF and IL-6. “Moonlighting” (a term originally used to describe a single protein with multiple functions that are not due to gene fusions or splice variants) may also apply to Cbl insofar as it has at least two functions: in addition to its canonical co-enzyme functions, it also modulates the synthesis of cytokines and growth factors in the CNS as well as in organs that are not morphologically affected by Cbl withdrawal (e.g. liver and kidneys) [2-4].
We then hypothesised that steps that stay behind the Cbl regulated cytokines and growth factors might be modified by Cbl deficiency, and further experiments demonstrated that: a) the Cbl deficiency-induced increases in TNF-α and NGF levels in the SC of Cbl-D rats are also responsible for increased SC levels of an activated transcription factor, nuclear factor(NF)-κB, which is capable of binding DNA; and b) Cbl up-regulates p75 neurotrophin receptor-immunoreactive cell levels and EGF receptor expression in the white matter of rat SC. As Cbl is classically considered a micronutrient, studies of NF-κB levels in Cbl-D rats can be included in the field of nutritional genomics or nutrigenomics. Furthermore, as Cbl indirectly regulates transcription processes by modulating NF-κB levels, Cbl has another mechanism for modulating DNA expression in addition to the regulation of methylation reactions [4, 5].
Cbl and PrPCs: A newly identified relationship between two key neurobiological molecules
Finally, we hypothesised that the normal cellular prion (PrPC) isoform may be indirectly regulated by Cbl in rat CNS, and therefore involved in the pathogenesis of rat Cbl-D neuropathy. The rationale underlying this hypothesis was that: a) PrPCs play a crucial role in maintaining CNS myelin; b) Cbl deficiency damages SC myelin by increasing TNF-α and NGF, and decreasing EGF levels; and c) other authors have shown that TNF-α and EGF regulate PrPC expression in vitro. The subsequent experiments demonstrated that: a) PrPC levels have increased in the SC and peripheral nerves (but not liver) of Cbl-D rats by the time myelin lesions appear; b) these increases are mediated by excess myelinotoxic TNF-α in Cbl-D SC and sciatic nerves, because the Cbl-D rats treated with i.c.v. microinjections of anti-TNF-α Abs showed no increase in SC, CSF and sciatic PrPC levels, and had normal SC and PNS myelin ultrastructures; c) Cbl and EGF up-regulate PrPC mRNA levels in rat SC and duodenal mucosa (but not liver), albeit to different degrees; d) repeated i.c.v. microinjections of Abs against the octapeptide repeat (OR) region of the PrPC molecule normalise SC and PNS myelin morphology, SC and PNS TNF-α levels, and maximum nerve conduction velocity (MNCV) in Cbl-D rats even if their Cbl-D status remains unchanged; and e) ON rats repeatedly treated with i.c.v. PrPC microinjections show spongy vacuolation in SC and myelin lesions in sciatic nerves, together with significantly reduced MNCV values. This shows that an excess of OR regions is involved in the pathogenesis of the CNS and PNS lesions observed in Cbl-D rats. It is believed that this is the first demonstration that an experimental myelin disease (rat Cbl-D neuropathy) can be caused by a local excess of PrPC OR regions. The number of OR regions in rat SC and peripheral nerves is therefore “buffered” by still unknown mechanisms in order to keep myelin normal, and Cbl plays a key role in this. In conclusion, rat Cbl-D neuropathy seems to be the first neurological disease shown to be due to excess PrPCs without any need for them to be conformationally transformed into a pathological form as in the case of PrP scrapie (PrPSC) [6,7].
Once again, we investigated whether PrPC increases similar to those observed in Cbl-D rats also occur in the CSF and serum of patients with clinically confirmed severe Cbl deficiency (i.e., with PA and/or SCD). Mean CSF PrPC levels in SCD patients were significantly higher than in the neurological controls; but no significant change was observed in patients with Alzheimer’s disease or amyotrophic lateral sclerosis in comparison with neurological controls. Mean serum PrPC levels in the PA patients were significantly higher than those in the controls, but normalized after Cbl therapy. There was no significant change in the sera of the patients with other forms of anemia [6,7]. As the serum and CSF PrPC levels in each of the groups of Cbl-D patients (PA and SCD) correlated significantly with serum and CSF Cbl levels, it can be concluded that Cbl regulates serum and CSF PrPC levels in these physiological fluids [6,7].
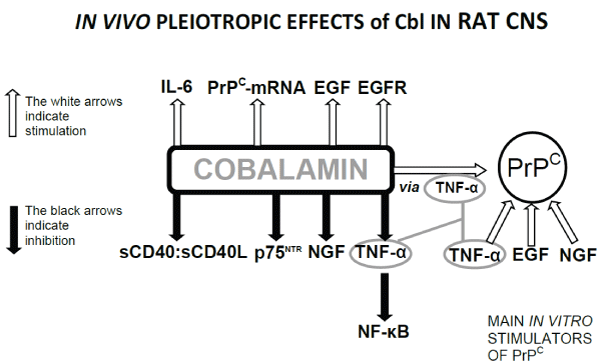
The most innovative results of the research led by Dr. Scalabrino are summarised in the figure below
References
- Scalabrino G(2001) Subacute combined degeneration one century later. The neurotrophic action of cobalamin (vitamin B12) revisited. J Neuropathol Exp Neurol 60: 109-120.
- Scalabrino G (2005) Cobalamin (vitamin B12) in subacute combined degeneration and beyond: traditional interpretation and novel theories. Exp Neurol 192: 463-479.
- Scalabrino G, Peracchi M (2006) New insights into the pathophysiology of cobalamin deficiency. Trends Mol Med 12: 247-254.
- Scalabrino G, Veber D, Mutti E (2008) Experimental and clinical evidence of the role of cytokines and growth factors in the pathogenesis of acquired cobalamin-deficient leukoneuropathy. Brain Res Rev 59: 42-54.
- Scalabrino G (2009)The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: Lessons learned from its deficiency. ProgNeurobiol88: 203-220.
- Scalabrino G, Veber D, Tredici G (2014) Relationships between cobalamin, epidermal growth factor, and normal prions in the myelin maintenance of central nervous system. Int J Biochem Cell Biol55: 232-241.
- Veber D, Scalabrino G (2015) Are PrPCs involved in some myelin diseases? Relating experimental studies to human pathology. J NeurolSci359: 396-403.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences